The sequence to be predicted must be specified in one of two ways:
As the DisEMBL prediction method makes use of sequence context, predictions on fragments will often give wrong results. One should thus always submit the full length sequence if at all possible.
DisEMBL has a six parameters that can be fine tuned by the user. Except from increasing the Savitzky-Golay smoothing frame for long proteins, these parameters should generally be left at their defaults.
Once you have specified the sequence (and possibly changed parameters) simply press the "DisEMBL protein" button to get the prediction results.
We describe protein disorder as two-state models where each residue is either ordered or disordered. For this purpose we used three different criteria for defining which residues are disordered:
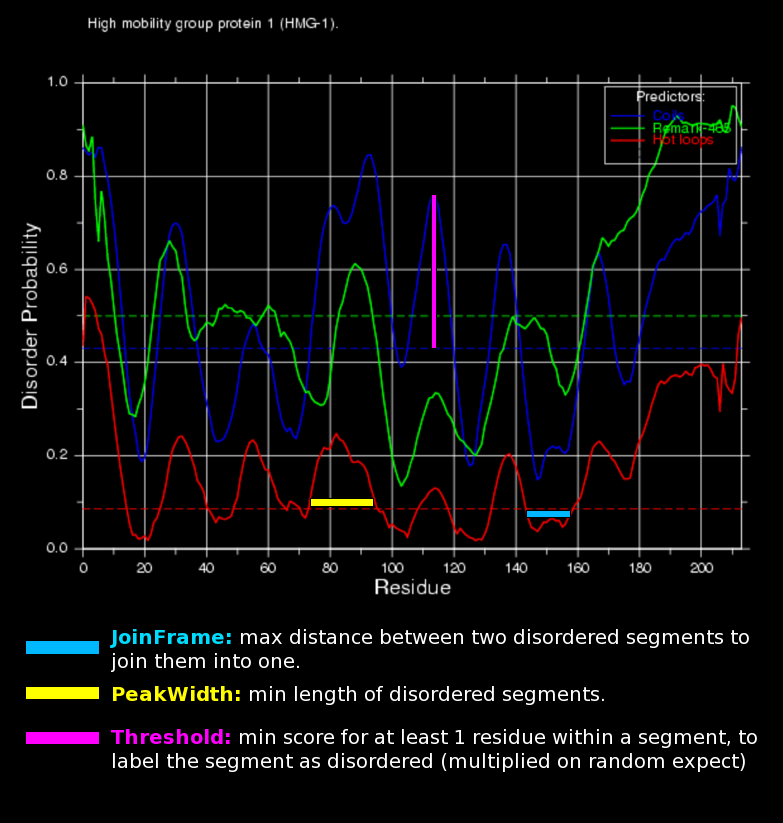
Predictions are shown according to each of the three definitions above. The predicted probabilities are shown as curves along the sequence and scores should always be compared to the corresponding random expectation value (dotted lines).
To interpret the predictions it is crucial to keep in mind that the three predictors are not all predicting the same kind of disorder. Agreement between the predictors should thus not be expected.
Certain sequence biased regions such as coil-coils might be predicted as disordered, which is to a certain extent true since these are belived to be structured in a globular manner only when they are bound to eachother. Again this illustrates that the capability of proteins to perform disorder-order transitions are ultimately and intrinsically described by their sequence!
If you want to run DisEMBL on large numbers of sequences, using the web interface is not the optimal solution. A much better option is to download the DisEMBL pipeline and install it locally. This allows DisEMBL to be deployed in a fully automated fashion on sequence files with arbitrarily many protein sequences.
The webinterface can also be used for large scale predictions, but please let us know if you want to do this. And always do it with moderation, i.e.
one job per 10 secs or so. The way to automate is by submitting jobs like this:
http://dis.embl.de/cgiDict.py?key=process&SP_entry=PRIO_HUMAN or http://dis.embl.de/cgiDict.py?key=process&sequence_string=SEQ
where SEQ and PRIO_HUMAN can be replaced.
DisEMBL is a public web server for predicting disorder in proteins. Although the GlobPlot server also predicts protein disorder, the two methods complement each other as they offer different approaches/features.
The web interface is fairly straight forward to use, the user can paste a sequence or enter the SWISS-PROT/SWALL accession (e.g.. P08630) or entry code (e.g. PRIO_HUMAN). DisEMBL fetches the sequence and description of the polypeptide from an ExPASy server using Biopython.org software.
The probability of disorder is shown graphically. The green curve is the predictions for missing coordinates, red for the hot loop network and blue for coil. The random expectation levels for the different predictors are shown on the graph as horizontal lines but should only be considered an absolute minima.
Normally the default parameters should not be changed. If the query protein sequence is very long, >1000 residues, you can download the predictions and use a local graph/plotting tool such as Grace or OpenOffice.org to plot and zoom the data. Having identified the potential disordered regions, you should now have a good basis for setting up expression vectors and/or comparing the data with obtained structural data. We encourage any feedback on success/failures in deploying DisEMBL in structural analysis of proteins!
The web server only allows predictions on one sequence at a time, if bulk predictions are needed we supply DisEMBL as a pipeline software package. The pipeline consists of the same three neural networks implemented as one C code module, which reads sequence from STDIN and writes predictions to STDOUT. The pipeline interface is intended for the structural genomics initiatives, it can analyse in the order of 1 million residues/min on a 1GHz 686 PC. This allows for very large scale predictions, e.g. as part of a structure space scanning. DisEMBL is released as OSI certified opensource software and can be downloaded here.
If you need further help please contact Rune Linding.